10 月 30 日,Bioconductor 3.10 正式发布,该版本的 bioc 共包括了 1822 个软件包,384 个实验数据包,952 个注释包和 27 个工作流程,基于 R 3.6.1。 其中,新的软件包有 93 个,新的数据包 15 个,新的注释包 2 个,大量的旧包都进行了一波更新。 接下来推荐几个值得关注的最新软件包,以及一些经典包的重要更新。 在 3.10 版本中新加入的 bioc 包,我个人比较感兴趣的有如下几个。 ALPS: AnaLysis routines for ePigenomicS data 一个最新的表观数据(ChIP-seq, ATAC-seq 等)可视化工具包,帮助你绘制高质量的发表级数据。下图是一个可以展示的内容概览。 输入数据是一个包含有 bigwig 源文件位置信息的 data table。再结合其它一些相关 R 包的处理结果可以展示各种类型的信息,例如计算基因组的富集区域供后续进行 PCA 和聚类的分析;绘制不同样品之间的富集差异;绘制基因浏览器或者 IGV 类似的 track 等等。同时也可以对基因组区域进行注释和绘制 motif。 作者也强调,这个包中的大多数图都可以利用 ggplot2 的一套系统进行二次定制。 在分析甲基化数据的时候比较关注的一个信息叫做 differentially methylated region (DMR) 差异甲基化区域,类似于转录组分析中的差异表达基因。 大多数已有的一些工具(其实也没有很多)针对的都是两组数据,而 MethCP 除了支持常规的两组比较外,也可以处理多种条件的同时比较,例如时序数据。 需要说明的是,MethCP 本身使用 bsseq 这个包加载原始数据。 目前甲基化的数据上游分析产生的基本都会是一个超大的 bedgraph 文件,一般性能的电脑基本是处理不了的。Methrix 这个工具包主要的设计目的就是处理这种大型的甲基化 bedgraph 文件。 在读入文件的同时,还可以利用参考基因组填补确实的 CpG 信息,并且创建甲基化矩阵。后续可以在矩阵的基础上进行一系列过滤和提取操作,并进行可视化展示。当然也可以把这个矩阵结果转换为 bsseq 数据包支持的格式,然后再使用 MethCP 这个包进行分析。 一个据介绍是可以直接完成从原始数据下载到完成所有常规分析的工具包。下图是主要分析流程。 在原始数据下载比对这个步骤中,其可以调用预编译好的 Bowtie2, Kallisto 和 salmon 的工具。首先可以利用 在进行 Biomarkers identification 的步骤时,可以进行质控和去除 batch effect 等操作,差异分析之后可以结合机器学习方法进行分类和基因筛选。 最后针对差异进行可已经常用的各种富集分析,包括 GO pathway 和 相关疾病。 (从个人经验来说,这种大而全的包质量一般不会特别好,但是可能还是会有一些人喜欢。) 在转录翻译的过程中,mRNA 会在尾巴处添加一些 ployA,这个 A 可是有讲究的,长短位置的不同都会都 mRNA 的稳定性带来各种各样的影响,于是有一个研究方向就是 APA (alternative polyadenylation)。 这方面研究相对权威的新泽西州罗格斯癌症研究所 Bin Tian 实验室针对人类中以后的可信 PolyA 位点信息,开发了一个使用转录组数据分析 APA 的工具包 APAlyzer。当前的版本支持检测 UTR 区域和内含子区域的 APA,并利用编码区进行表达分析。 大量的已有工具包在 3.10 版本中进行了更新,这里挑选几个大家比较熟悉的进行简要介绍。 ChIPseeker 是 Y 叔开发的一个对 genomic region 进行各种注释可视化展示的工具包。其中有一个图比较典型,如下所示,就是把 upsetplot 嵌入 vennpie。这个图在实现效果前前后后经历过几次改变,但是在这个包中的实现方法一直没有升级,近期这个图在使用过程中偶尔会出现一些问题,例如只显示饼图不显示 upsetplot 等,然后 Y 叔就把这个实现方法给升级了。 用 Y 叔的原话说就是 于是我就把 ChIPseeker::upsetplo t 重新给实现了,利用了 ggimage + ggplotify,代码长度变成原来的 1/3,而且不会出现上面的这些问题。 之前写过一篇文章介绍 船新版本 DESeq2 处理大量样本速度显著提升,现在这个版本的 DESeq2 也随着 bioc 的升级而正式升级。 在之前的文章中,已经比较详细的写了为什么在处理大量样本时速度会有急速提升。在升级说明中则写的比较含蓄 speeds up DESeq2 for large sample sizes (n > 100) by at least an order of magnitude. In fact the speed is now linear with number of samples whereas previously DESeq2 would scale quadratically. 调用 Ensembl Variant Effect Predictor 的 perl API 进行突变注释分析的 R 包 ensemblVEP 现在支持了 Ensembl release 97/98。似乎没啥可说,但是还是挺重要的。因为每一次 Ensembl 的更新,在一些物种上都会有比较大的升级。 IsoformSwitchAnalyzeR 是一个可以鉴定,注释和可视化可变剪切和转录本转换的工具包。在 3.10 中进行了大量的升级,多数函数都有改变。 maftools 是分析和展示 Mutation Annotation Format (MAF) 文件的工具包,在不少文章中都可以这个包绘制的图。在这次更新中,maftools 增加了 survGroup, mafSurvGroup 两个函数,用来预测和生存相关的基因以及基因集。另外,Signature analysis 分析步骤也有很多调整。 其它具体的更新信息,可以参考官方说明。 查看当前版本: 应该会展示如下信息 绝大多数情况下升级只需要执行如下命令: 通过指定版本号之后,所有可以更新的 R 包都会更新到 3.10 版本。如果你安装的 R 包比较多,提示需要更新一两百个 R 包都是很正常的。祝好~ 本文作者:思考问题的熊 版权声明:本博客所有文章除特别声明外,均采用 知识共享署名-非商业性使用-禁止演绎 4.0 国际许可协议 (CC BY-NC-ND 4.0) 进行许可。 如果你对这篇文章感兴趣,欢迎通过邮箱订阅我的 「熊言熊语」会员通讯,我将第一时间与你分享肿瘤生物医药领域最新行业研究进展和我的所思所学所想,点此链接即可进行免费订阅。新加入的 bioc 包
ALPS

MethCP & Methrix
Knowseq
downloadPublicSeries(c("GSE74251")) 下载 GEO 中的数据信息,然后使用read.csv("ReferenceFiles/GSE74251.csv") 读取文件中的数据信息,接下来就可以使用 rawAlignment 这个命令来进行序列比对了。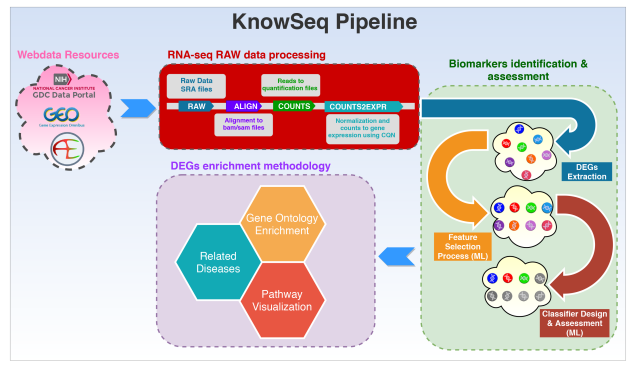
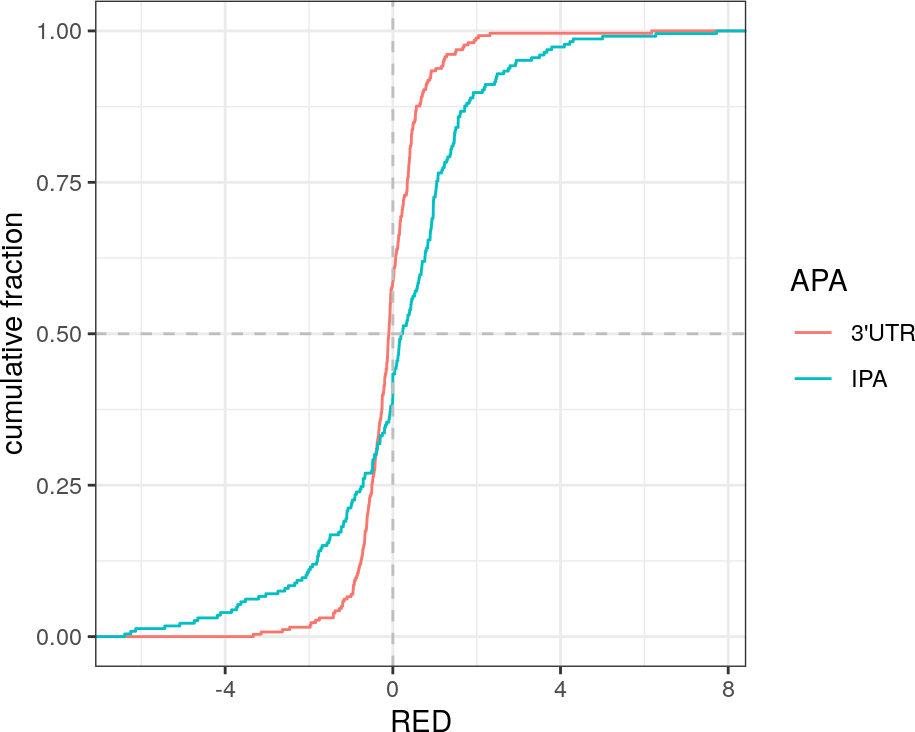
APAlyzer
有重要更新的 R 包
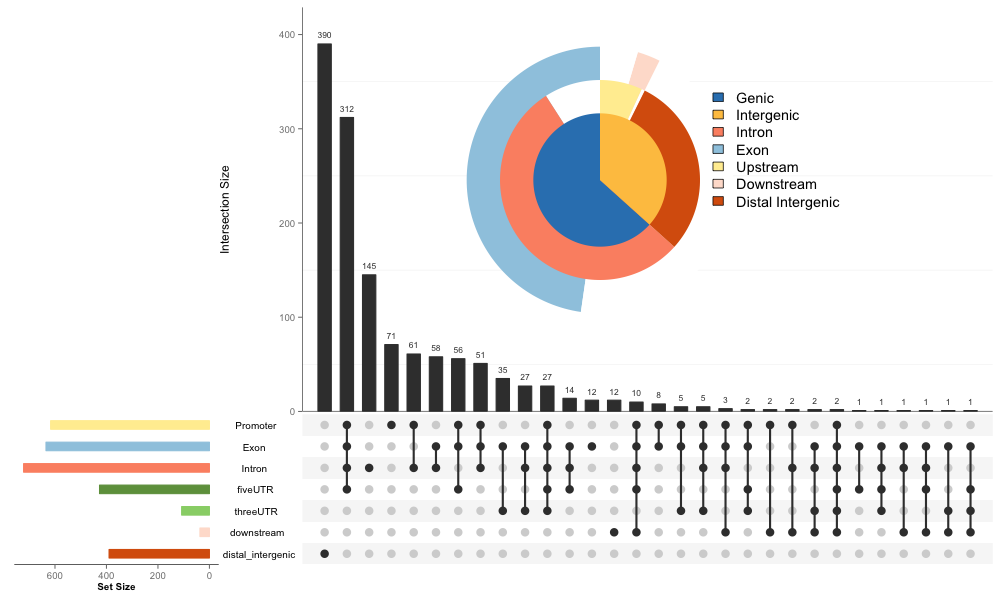
ChIPseeker
library(ChIPseeker)
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene
library(clusterProfiler)
files <- getSampleFiles()
print(files)
peakAnno <- annotatePeak(files[[4]], tssRegion=c(-3000, 3000),
TxDb=txdb)
upsetplot(peakAnno, vennpie=T)
DESeq2
ensemblVEP
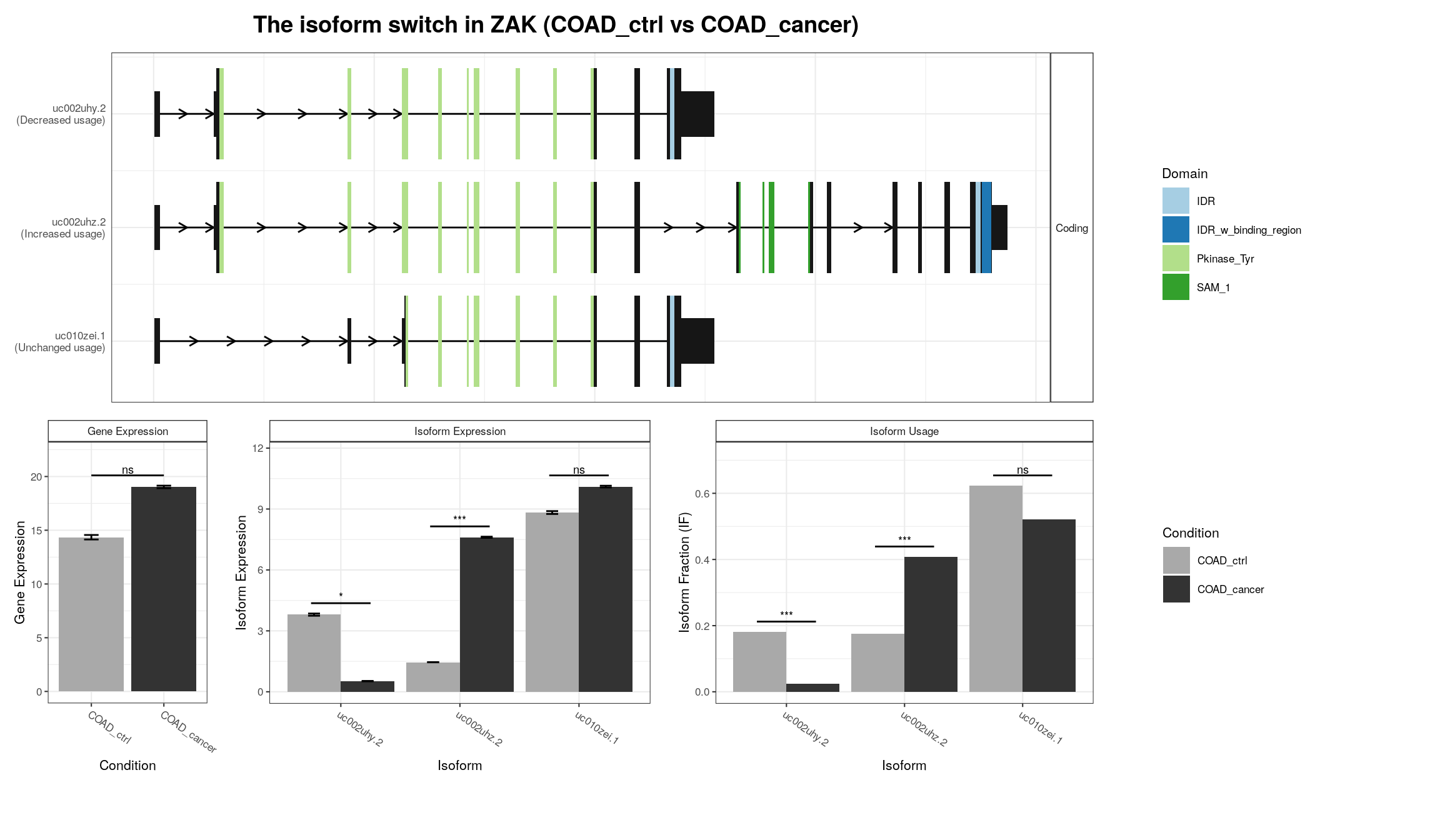
IsoformSwitchAnalyzeR
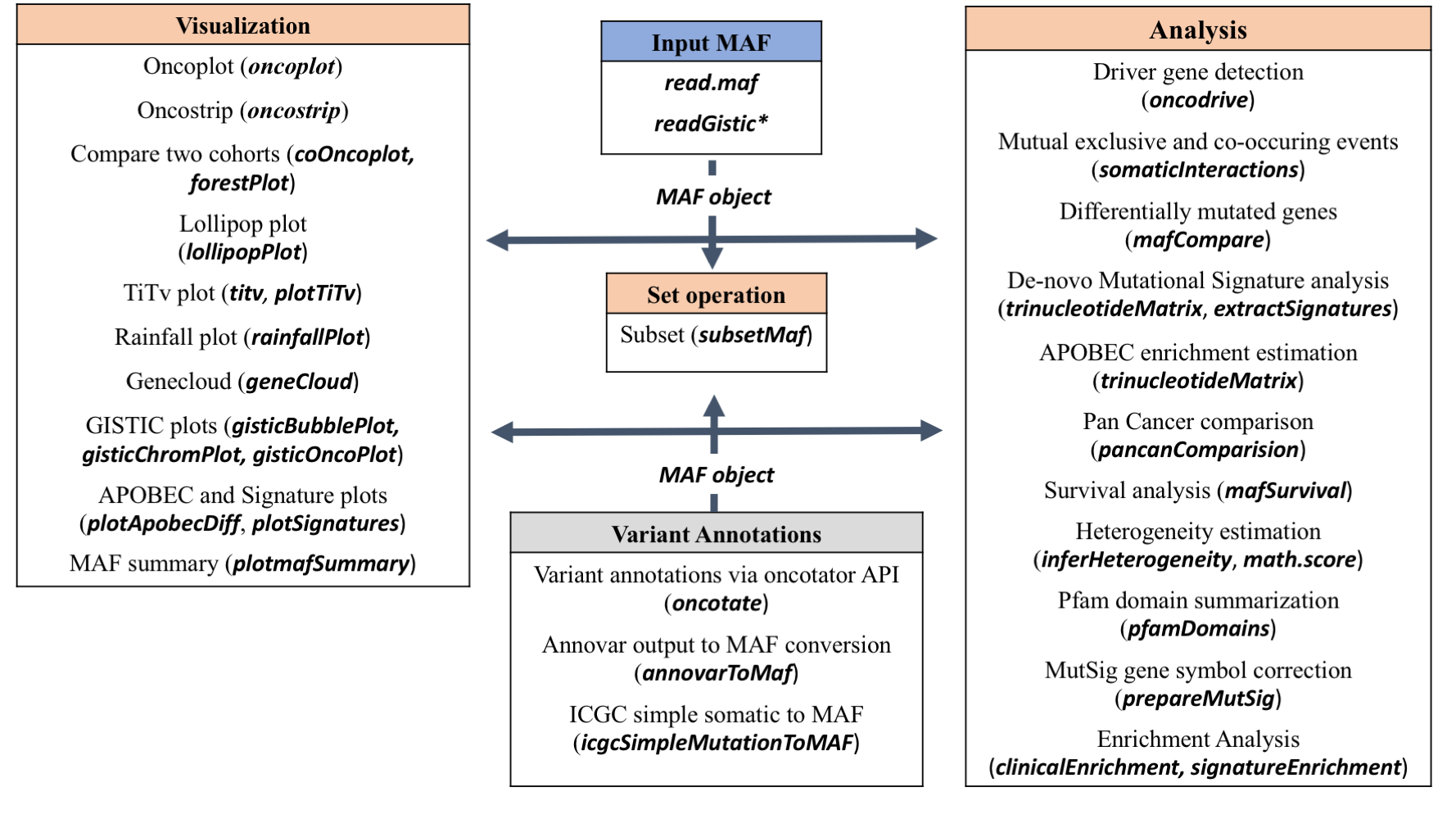
maftools
如何升级
library(BiocManager)
Bioconductor version 3.9 (BiocManager 1.30.4), ?BiocManager::install
for help
BiocManager::install(version = "3.10")
· 分享链接 https://kaopubear.top/blog/2019-10-31-bioconductor310iscoming/